Buscan identificar de manera minuciosa las variaciones genéticas que pueden influir en la predisposición o desarrollo de la esofagitis eosinofílica.

El pasado 5 de enero del 2024 se difundió un paso crucial en la comprensión de la esofagitis eosinofílica (EoE), un trastorno atópico poco común asociado con problemas esofágicos y síntomas como dificultad para tragar e inflamación, por medio de un estudio de la revista AJHG, en el que se destaca que la EoE se desarrolla en un pequeño subconjunto de personas con alergias alimentarias.
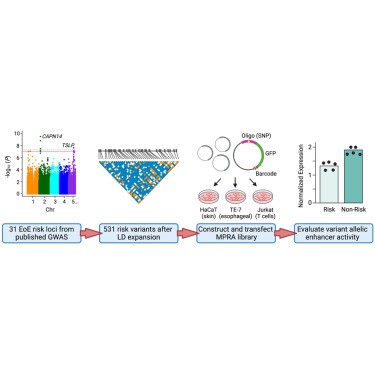
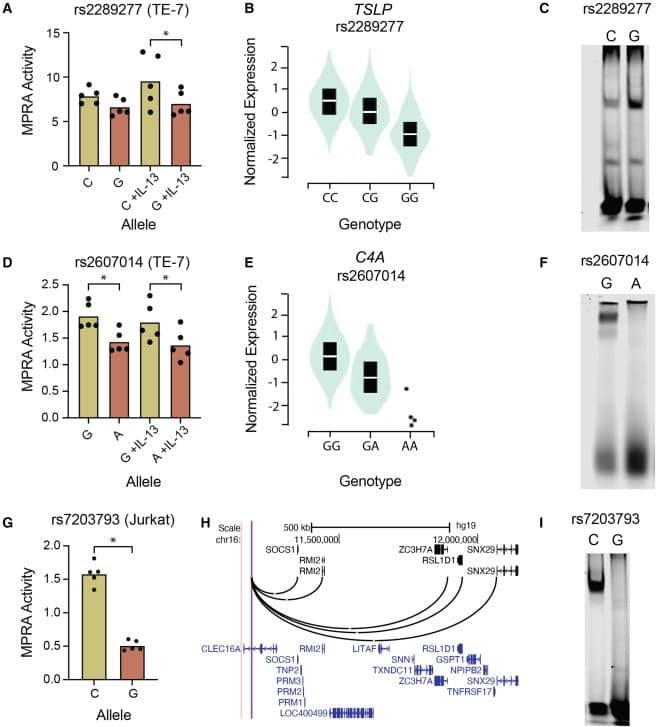
Utilizando estudios de asociación de todo el genoma (GWAS), los científicos identificaron 9 locus de riesgo independientes de EoE y 27 loci adicionales de importancia sugestiva. En el último estudio, realizaron una expansión del desequilibrio de ligamiento (LD) de estos loci para nominar un conjunto de 531 variantes que podrían ser potencialmente causales.
Tal avance brinda una visión más detallada de las variaciones genéticas relacionadas con la EoE, allanando el camino para futuras investigaciones y posibles avances en el diagnóstico y tratamiento de esta condición.
En este estudio se seleccionaron y generaron secuencias de ADN relacionadas con los lugares del genoma vinculados a esta enfermedad, EoE. Utilizaron datos genéticos hasta abril de 2019 y 41 variantes representativas de 9 lugares genéticos de riesgo significativos y 27 lugares sugerentes de riesgo. Luego, expandieron estas ubicaciones utilizando datos genómicos y diseñaron secuencias de ADN para cada variante, estas secuencias se depositaron en un banco de datos, y se analizaron específicamente las variantes asociadas con la EE.
La primera se hizo según un protocolo establecido y se usó en experimentos relacionados con TE-7. Para la segunda versión, agregaron un paso extra: después de que la biblioteca principal se transformó en Escherichia coli (E. coli), se dividió en varias porciones y se recuperaron. Con esto, realizaron estimaciones del número de unidades formadoras de colonias y luego se expandieron volúmenes suficientes para diferentes multiplicadores del número de alelos en el conjunto de oligos.
Luego de extraer los plásmidos, amplificaron las regiones oligo y de código de barras para construir bibliotecas de secuenciación, esas bibliotecas se purificaron, combinaron y se secuenciaron utilizando tecnología Illumina NovaSeq 6000. Después del análisis, eligieron una biblioteca principal con 211 códigos de barras únicos medianos para construir la biblioteca de transfección. La versión que mencionan, se empleó en experimentos relacionados con HaCaT y Jurkat.
En cuanto al cultivo celular y transfección, las líneas celulares HaCaT, TE-7 y Jurkat fueron cultivadas en medios específicos con suplementos; luego, ejecutaron transfecciones replicadas utilizando plásmidos de biblioteca de transfección y reactivos según las instrucciones del fabricante. Después de la transfección, las células se recuperaron en medios adecuados durante 24 horas y se estimularon con sustancias específicas. Posteriormente, recogieron las células para la preparación de la biblioteca de secuenciación y el conteo de códigos de barras. Proceso que se llevó a cabo de manera afín en las tres líneas celulares mencionadas.
En el ciclo que se construía de la biblioteca de secuenciación para el recuento de códigos de barras, “Se extrajo el ARN total usando un kit específico y se trató con ADNasa, se realizó el enriquecimiento y elución del ARNm de eGFP utilizando sondas biotiniladas, y el ARNm se transcribió a ADNc utilizando un sistema de síntesis de primera cadena”, aseguran los investigadores.
Las concentraciones relativas de eGFP se estimaron mediante PCR y las muestras se diluyeron para igualarlas, realizaron dos PCR para construir la biblioteca de secuenciación, y después de una amplificación adicional, las muestras fueron purificadas, agrupadas por morales y se secuenciaron utilizando el sistema NovaSeq 6000.
En la asociación de oligo y códigos de barras, las lecturas de extremo emparejado de 125 pb fueron filtradas y procesadas, por lo que la lectura 1 se dividió en la región de código de barras y la región de “oligocoincidencia”, y ambas regiones se mapearon a las secuencias de oligos sintetizadas; los códigos de barras se asociaron con las secuencias de oligo utilizando el ID de lectura, y sólo se utilizaron los códigos de barras asignados de manera única para análisis posteriores.
Una vez se halló el dato anterior procedieron con el conteo de códigos de barras, seleccionaron cuidadosamente lecturas de 100 pb asegurándose de su calidad, cada lectura se dividió en la región del código de barras y la región constante, siendo esta última emparejada con la UTR 3' de eGFP. Revelaron que sólo se conservaron las lecturas que cumplían con requisitos específicos, como una coincidencia precisa en la región constante y adyacencia al código de barras.
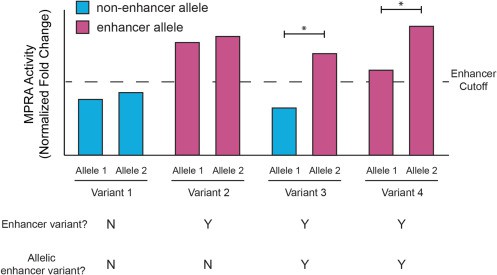
Fue allí cuando iniciaron la búsqueda de variantes con actividad potenciadora, analizaron los oligos con muchos códigos de barras únicos del control de plásmido, compararon los controles de plásmido con las réplicas experimentales; con el fin de etiquetar una variante como potenciadora siempre y cuando mostrara actividad en al menos un alelo.
Tras lo anterior, extrajeron el ARN total de células estimuladas con IL-13 o agua (control) utilizando un kit de aislamiento específico; Anotaron y analizaron genes diana usando información de interacciones de bucles de ADN y datos eQTL del catálogo eQTL y GTEx. Luego, a los genes asociados a eQTL los enriquecieron en procesos biológicos según la ontología genética (GO).
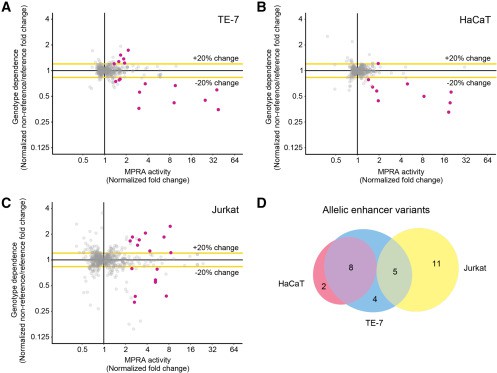
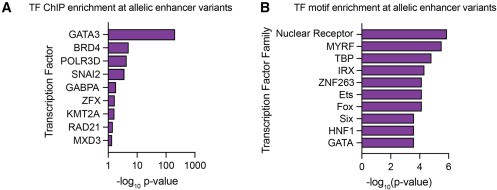
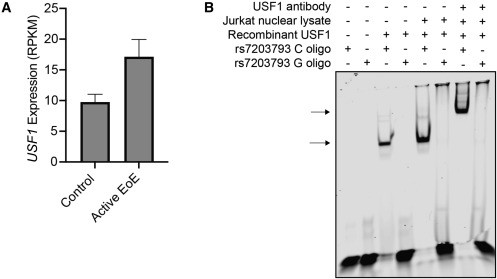
Se Identificaron 32 variantes genéticas de riesgo que alteran la expresión genética en células esofágicas, cutáneas y de células T; entre estas variantes se destacaron GATA3 y USF1, factores de transcripción asociados a la inflamación alérgica y sobreexpresados en pacientes con EEo.
Estos hallazgos abren la puerta a estrategias de medicina personalizada para diagnosticar y tratar la enfermedad de manera más efectiva, esto con la herramienta nombrada ensayos de reporteros masivos en paralelo (MPRA).
Para leer la fuente, haga clic aquí.